Understanding Sortase A Catalysis
The Sortase enzymes are a ubiquitous and highly conserved family of transpeptidases that catalyze the incorporation of cell surface proteins into the prokaryotic cell wall. The Staphylococcus aureus, Sortase A (SrtA) plays an essential for in bacterial pathogenesis and therefore, has been the primary focus of mechanistic and structural investigations for therapeutic development. However, more recently SrtA has received attention for its potential use as a molecular reagent to synthesize novel bio-conjugates. SrtA covalently couples surface proteins containing the carboxy-terminal pentapeptide recognition motif, or sort-tag (LP-X-TG, where X represents any amino acid), to peptidoglycan precursors on the exterior plasma membrane. In an elegant kinetic analysis, Frankel and colleagues revealed that SrtA utilizes a reverse-protonation mechanism in which the Cys184 and His120 side chains are deprotonated and protonated, respectively during catalysis. Therefore, at the pH optimum for acyl formation (pH 7.0-8.5), only a fraction of the total enzyme is in an active or competent state for catalysis.
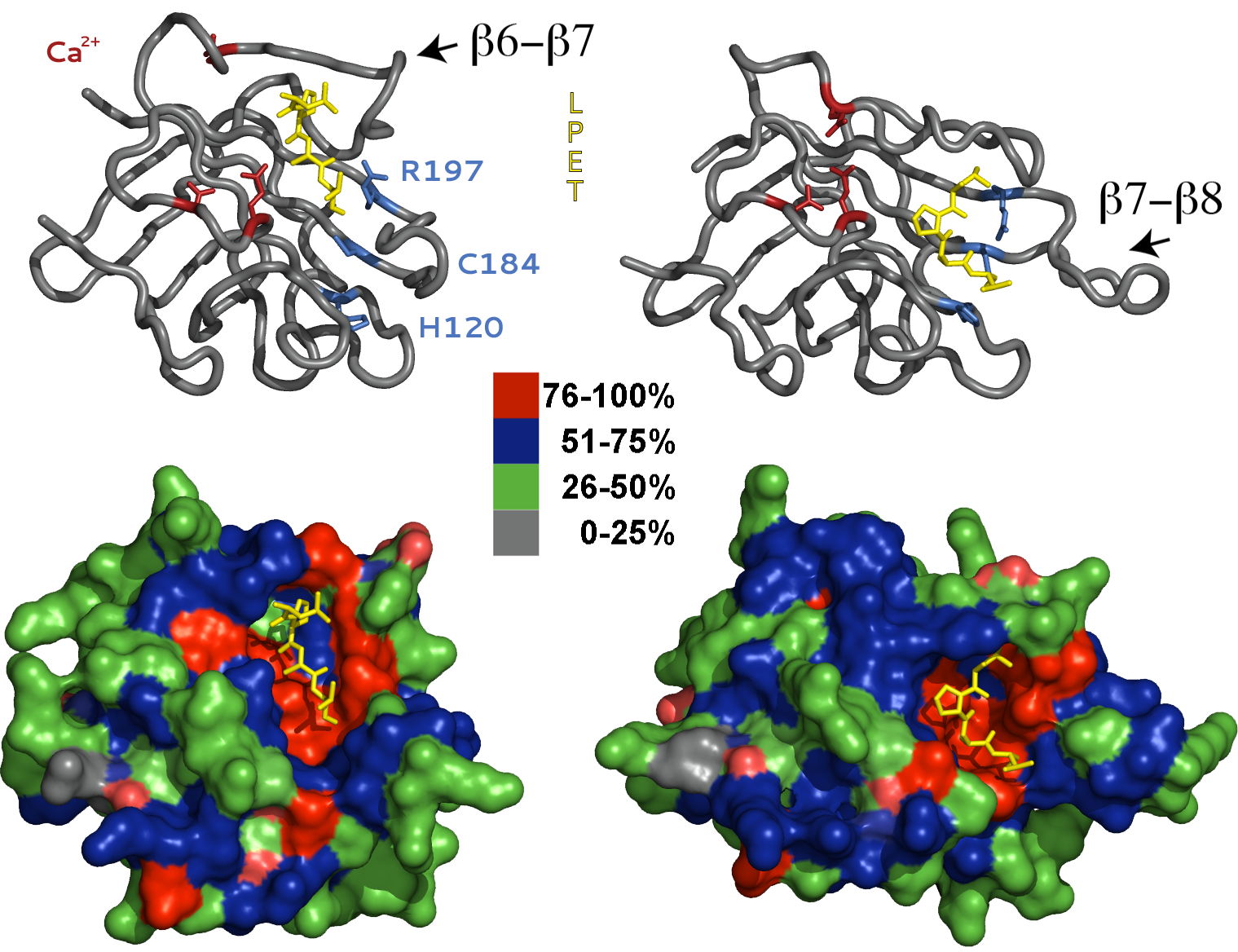
NMR and x-ray structures demonstrate that SrtA adopts an eight-stranded β-barrel fold, which is well conserved among other Sortase family members including SrtB and SrtC. However, the two models differ significantly in the orientation of the sort-tag substrate and positioning of the β6-β7 and β7-β8 loops. In the crystal structure, the LPETG substrate is bound to a shallow solvent exposed crevice several angstroms from the active-site pocket. In addition, the key catalytic residues, Cys184 and His120, are improperly oriented and positioned for catalysis. Alternatively, in the NMR model the sort-tag is positioned in a deep groove created by opening of the β7-β8 loop, and residues Cys184 and His120 are properly aligned for thioester formation. In light of these discrepancies, it is widely regarded in the literature that substrate binding in the crystal structure is non-specific!
In contrast, we postulate that the two models represent distinct structural conformations adopted by SrtA during catalysis. We believe the x-ray model represents the initial substrate-binding event. Once bound, the enzyme undergoes a dramatic conformational change that simultaneously opens the active-site pocket and repositions and orients Cys184 and His120 for catalysis. In addition, rotation and closing of the β6-β7 loop and separation of the β7-β8 loop from the protein core translates the sort-tag substrate to the newly formed active site pocket. The catalytic residues Cys184 and His120 also rotate and approach one another and adopt positions appropriate for catalysis. Our model is consistent with the reverse-protonation catalytic mechanism and provides a logical explanation for SrtA mutations that enhance catalysis despite their significant distance from the active site pocket. Dynamic conformational changes have been implied in several recent publications, however, direct experimental evidence is still lacking.