Mammalian Cell Expression
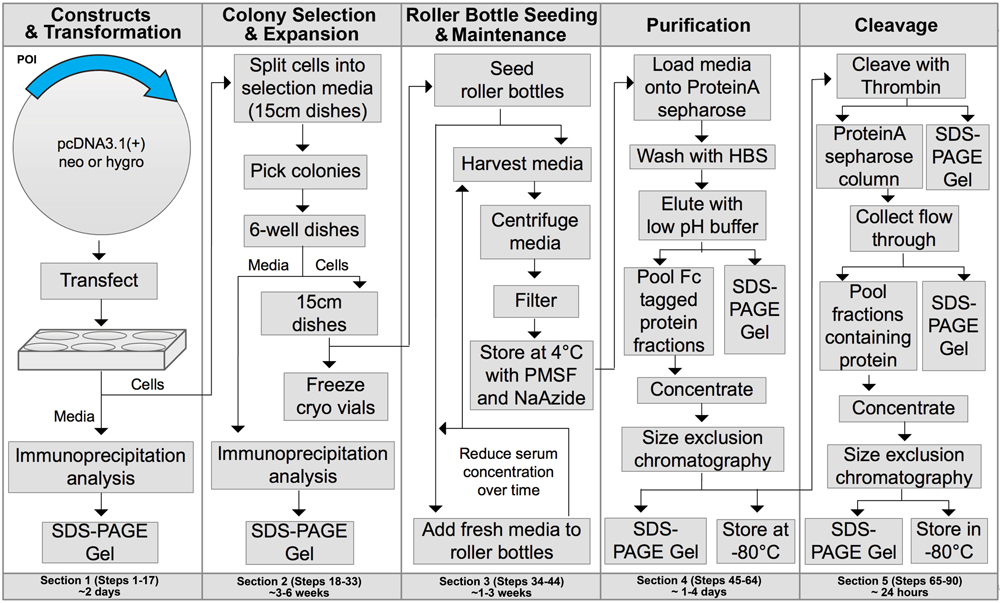
A few protocols for working with mammalian cells:
Cultivation of Human Embryonic Kidney 293 Cells Medium: 500 mL Dulbecco's Modified Eagle (DME-HG) 50 mL Fetal Bovine Serum (FBS) - 10 % of the medium volume 5 mL penicillin/streptomycin (10 000U/mL Pn and 10 mg/mL Str)-1x final concentration Trypsin (0.05% solution with 0.5 mM EDTA) Phosphate Saline Buffer (PBS), pH 7.0-7.2 Procedures: Starting from a Cryotube: Prewarm the medium at 37°C and fill one 15 cm dish with 25 mL. DO NOT USE MEDIA THAT CONTAINS HYGRO. , G418, etc. AT THIS POINT! Rapidly thaw the cells in your hand, gently pippet up and down to thaw any remaining ice before distributing in a dish filled with warm media. Rock the dish to distribute the cells. Do not swirl-if you do they will all clump towards the center of the plate. Change the medium after 24 hrs or once the cells have attached. Splitting Cells (all solutions must be prewarmed): Prepare a new dish with fresh medium. (25 mL for 15 cm plates, 10 mL for 10 cm plates) Aspirate the old medium from the dish. Wash the cells carefully with PBS to remove residual medium and FBS which can inhibit the trypsin. Trypsinize cells by adding 3-5 mL of trypsin to the dish and incubate at 37°C (or RT, it depends on its activity) until cells have detached (usually 2-5 minutes). Take a fraction of the cell solution and transfer it to the new dish (BSA in medium inhibits the trypsin). When splitting confluent 293 cells in a 1:10 ratio, confluence is reached again after 3-5 days.
Cultivation of HEK 293 in Roller Bottles Medium: 500 mL Dulbecco's Modified Eagle (DME-HG) 50 mL Fetal Bovine Serum (FBS) - 10 % of the medium volume 5 mL penicillin/streptomycin (10 000U/mL Pn and 10 mg/mL Str)-1x final concentration Trypsin (0.05% solution with 0.5 mM EDTA) Phosphate Saline Buffer (PBS), pH 7.0-7.2 ***The roller bottles used are ribbed, with ~ 1700-2200 cm2 of surface area. If you want to use bottles with 2200 or more cm2 surface area-you must increase the amount of cells you innoculate each bottle with. This is very important if you wish to avoid cell patches that will degrade the quality of your culture. Procedures: Aspirate the media (it might also be harvested in order to collect your protein). Pour directly in each roller bottle 350 mL of prewarmed media. Place the roller bottles in the incubator and let them roll at 0.1 rpm to warm up. Wash each 15 cm plate with approximately 10 mL PBS. Trypsinize the cells with 5 mL trypsin ( do not trypsinize more than 5 plates at a time). Pipette the suspension from ONE plate into ONE roller bottle. Put the roller bottles in the incubator and let sit overnight at 0.1 rpm to allow the cells to attach. When the cells are attached onto the bottles' surface (After ~ approximately 24h), increase the rotation to ~1rpm. Change the media once the cells reach confluence. At this point we usually change into serum free media (if that's what is called for), or just add fresh media and continue the culture. ***If using serum free media-after the first harvest (or once the cells reach confluence) wash each bottle once with ~50 mL serum-free media. After washing, add back app. 250-300 mL of media lacking serum. Harvest the serum free media after 2 days. Repeat if the cells appear healthy.